Music links
Music Tutorial - The pmap control file
The first thing that we need to do, is to create a potential map which stores the potential energy of the
interaction between a single sorbate sphere (e.g. CH4 for methane or CH3 for ethane) and the sorbent (IRMOF1)
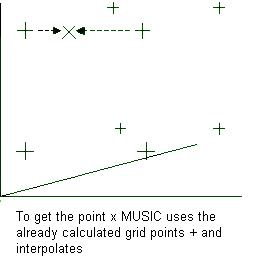
unit cell in a grid. When the GCMC simulation is run it uses this map to save time as it only needs to
interpolate the potentials between the grid points instead of calculating it from scratch.
An example file for downloading: MAP CONTROL FILE
The control files can be found in the music/ctrlfiles directory. Copy "music.makemap" into the folder where you
want to run the simulation. You have to customise the example file found in the ctrlfiles directory for your own
needs. The following shows the file modified to get a potential map for methane in IRMOF1. The text in red shows
helpfull tips, the actual control file has extra comments explaining the meaning of most input variables.
------ General Information ------------------------------------------
Methane in IRMOF1 some info so you can tell what the file simulates
1
1
1
1
2
1892134
4
Methane.res
Methane.con
------ Atomic Types --------------------------------------------------
5 number of atoms in your system
list all the atoms and their corresponding atom file
Methane methane here is an atom as it is modeled as a solid sphere
Methane.atm This is the atom file you created earlier
Carbon list all the atoms that make up the IRMOF1 framework
Carbon.atm
Oxygen
Oxygen.atm
Hydrogen
Hydrogen.atm
Zinc
Zinc.atm
------ Molecule Types -------------------------------------------------
2 Same again with the molecules
Methane
Methane.mol This is the molecule file you created earlier
IRMOF1
IRMOF1.mol
------ Simulation Cell Information --------------------------------------
IRMOF1 Name of your sorbent
2, 2, 3 how to replicate the unit cell in x,y,z directions
1, 1, 1
------ Forcefield Information -------------------------------------------
BASIC BASIC will use bruteforce calculations
MOL
A-A-inter List the input files that you created earlier - atom atom file
M-M-inter - molecule molecule file
intra.interactions - intramolecular interaction file
------ Mapmaker Information --------------------------------------------
1
DON'T REMOVE THIS LINE. Don't ask why, just don't.
IRMOF1 name of the sorbent
Methane name of your sorbate
NCOUL LJ Type of interaction to be mapped
0.2 Grid spacing
100.0 Potential cut off when overlap with framework, if U > 100., no value will be stored
AUTO Music can automatically create a filename or you can specify it explicitly.
Be aware that the filename shouldn't contain more than 29 letters!
------ Configuration Initialization -------------------------------------
Methane molecule name
Molecule NULL
IRMOF1
Fixed NULL