Our research in a nutshell
This video is part of the Research in a Nutshell initiative of the University of Edinburgh.


We use computer simulation to gain molecular-level insight into adsorption
and diffusion phenomena in nanoporous solids such as metal-organic
frameworks, zeolites and mesoporous oxides. Using molecular simulation we
can predict macroscopic adsorption properties such as the uptake of a
gas or the mixture selectivity (a measure of how well a solid discriminates
between different components in a mixture). More importantly, we also get
a detailed picture on the molecular scale which is not easily accessible
with experimental methods.
Molecular simulation helps us to understand the fundamentals and to asses
which molecular-level properties are responsible for the performance of a
porous solid. This insight is invaluable for finding promising materials
for a particular application and ultimately can help to develop better
materials. Molecular simulation also works hand-in-hand with experiments
to characterise porous materials and to understand what is going on. Our
work involves close collaboration with material chemists synthesising
porous materials as well as scientists and engineers interested in their
application.
This video is part of the Research in a Nutshell initiative of the University of Edinburgh.
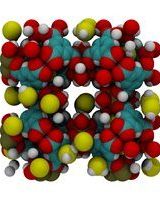
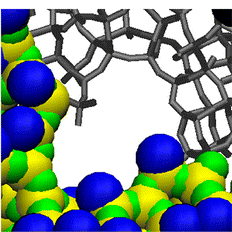
An ab initio parameterised force field yields correct description of adsorption on open metal sites.
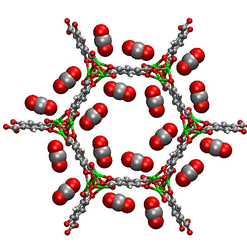
Packing effects play an important role in chiral separation of diols in a chiral MOFs.
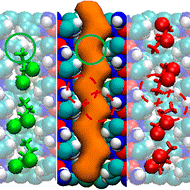
kMC simulation of SBA-2 synthesis reveals origin of connecting windows.