Research Links
Research
We use molecular simulation techniques to design innovative porous materials with properties tailored for specific adsorption applications. We are looking at a wide range of applications from carbon capture and hydrogen purification to liquid phase adsorption, nanomedicine and heterogeneous catalysis. Reflecting the interdisciplinary nature of the research, we are collaborating with researchers across the world with a wide variety of expertise ranging from material chemists synthesising porous materials to engineers interested in their applications. Given below are some examples of what we do.
We are interested in (adsorption) applications where molecular simulation provides insight on how the molecular level characteristics influence the macroscopic performance of a material.
Multiscale simulation of hydrogen purification using MOFs
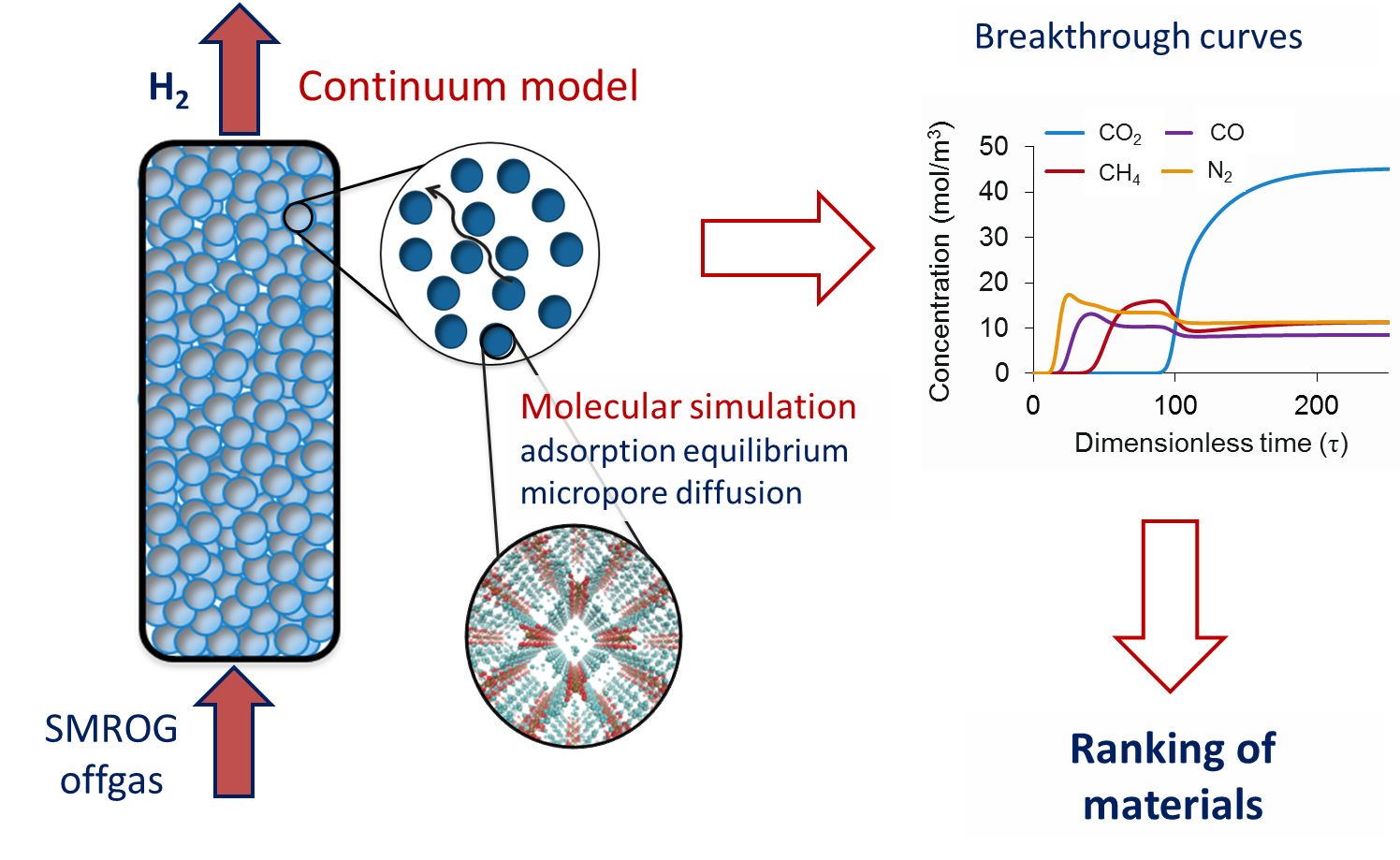 In order to assess the suitability of MOFs for hydrogen purification, we have identified a number of promising MOFs
using Monte Carlo and molecular dynamics simulations to predict adsorption isotherms, selectivities and micropore diffusion
coefficients. These molecular simulation results were then integrated into a full scale simulation of a pressure swing adsorption
column developed by Daniel Friedrich and Stefano Brandani. This multiscale simulation study identified promising materials
that outperform commercial zeolites studied in the literature for hydrogen purification from steam methane reformer offgas (SMROG).
In order to assess the suitability of MOFs for hydrogen purification, we have identified a number of promising MOFs
using Monte Carlo and molecular dynamics simulations to predict adsorption isotherms, selectivities and micropore diffusion
coefficients. These molecular simulation results were then integrated into a full scale simulation of a pressure swing adsorption
column developed by Daniel Friedrich and Stefano Brandani. This multiscale simulation study identified promising materials
that outperform commercial zeolites studied in the literature for hydrogen purification from steam methane reformer offgas (SMROG).
Exploiting open metal sites for carbon capture and nanomedicine
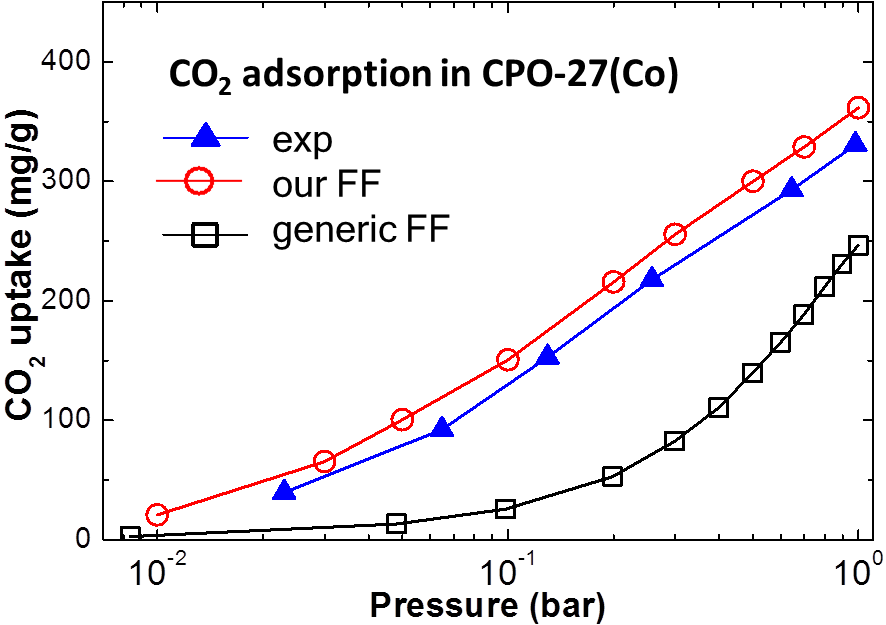 The most promising MOFs for carbon capture contain open metal sites.
However, as the interaction of CO2 with the open metal sites is considerably enhanced and is stronger than what is traditionally
considered as physisorption, conventional force fields often result in significant
underprediction of the uptake. We have recently developed methods that combine quantum chemical simulations with classical molecular
simulations to correctly describe these interactions and the predicted adsorption isotherms are in excellent agreement with
experimental results. Importantly, our force field parameters are transferable to other MOFs.
Open metal sites in MOFs not only play an important role
in the uptake of CO2 but also for the uptake of water and biologically active molecules such as CO and NO.
This behaviour is exploited for drug delivery where the more strongly interacting water molecules are displacing
NO molecules from the open metal sites thus enabling the controlled release of NO.
The most promising MOFs for carbon capture contain open metal sites.
However, as the interaction of CO2 with the open metal sites is considerably enhanced and is stronger than what is traditionally
considered as physisorption, conventional force fields often result in significant
underprediction of the uptake. We have recently developed methods that combine quantum chemical simulations with classical molecular
simulations to correctly describe these interactions and the predicted adsorption isotherms are in excellent agreement with
experimental results. Importantly, our force field parameters are transferable to other MOFs.
Open metal sites in MOFs not only play an important role
in the uptake of CO2 but also for the uptake of water and biologically active molecules such as CO and NO.
This behaviour is exploited for drug delivery where the more strongly interacting water molecules are displacing
NO molecules from the open metal sites thus enabling the controlled release of NO.
Many porous solids exhibit different degrees of flexibility upon guest inclusion ranging from large breathing movements observed in some MOFs to more subtle framework changes in MOFs and zeolites which nevertheless can have a dramatic influence on the adsorption behaviour and performance of these materials.
Gating
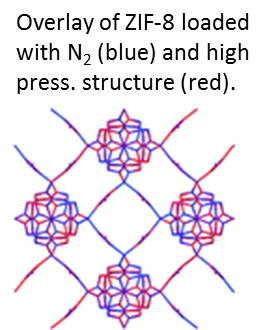 ZIF-8 is a MOF consisting of cavities interconnectd by windows. Because of the small size of the windows, it should allow
for molecular sieving of gases such as H2 and CH4. However, experimentally much larger molecules are taken up.
Through a combination of a variety of simulation and experimental techniques, we showed that the guest molecules are causing a
swing of the imidazolate linkers which opens the gate for much larger molecules. Interestingly, this gate opening
by gas uptake at low pressure is the same as observed in the empty structure at very high pressure (14,700 bar). While the
flexibility in ZIF-8 is detrimental to separation by adsorption, other examples exist where a small degree of framework
flexibility or distortion enhances the separation performance.
ZIF-8 is a MOF consisting of cavities interconnectd by windows. Because of the small size of the windows, it should allow
for molecular sieving of gases such as H2 and CH4. However, experimentally much larger molecules are taken up.
Through a combination of a variety of simulation and experimental techniques, we showed that the guest molecules are causing a
swing of the imidazolate linkers which opens the gate for much larger molecules. Interestingly, this gate opening
by gas uptake at low pressure is the same as observed in the empty structure at very high pressure (14,700 bar). While the
flexibility in ZIF-8 is detrimental to separation by adsorption, other examples exist where a small degree of framework
flexibility or distortion enhances the separation performance.
Breathing
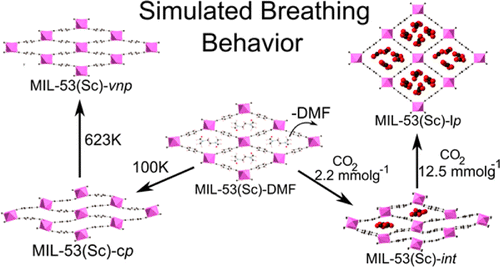 Some MOFs also show dramatic structural changes due to external stimuli as in the case of MIL-53. Our simulations allowed
for the first time to describe the significant structural changes of a metal-organic framework due to external stimuli from
first principles and without experimental input. Excellent agreement with experimental results was achieved and we have proven
that ab initio molecular dynamics simulations are an invaluable technique for understanding the origins of the structural
changes and structure determination when conventional methods fail.
Some MOFs also show dramatic structural changes due to external stimuli as in the case of MIL-53. Our simulations allowed
for the first time to describe the significant structural changes of a metal-organic framework due to external stimuli from
first principles and without experimental input. Excellent agreement with experimental results was achieved and we have proven
that ab initio molecular dynamics simulations are an invaluable technique for understanding the origins of the structural
changes and structure determination when conventional methods fail.
We are interested in different aspects of modelling synthesis and self-assembly processes of porous solids. The aim of this work is not only to create realistic models of the solids as input for the simulation of e.g. adsorption applications but also to gain a better understanding of how materials are formed and how the synthesis conditions influence the creation of particular structures.
MOF synthesis
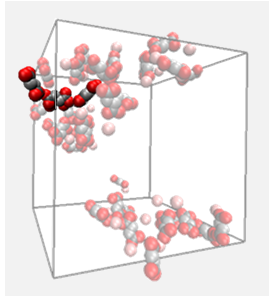 MOFs are synthesised in a self-assembly process from different building blocks: metal corners and organic linkers are combined
in a kind of molecular Lego set. This modular synthesis leads to a near infinite number of possible structures. Depending on
the synthesis conditions different structures can be synthesised even from the same building blocks making it difficult to target
a specific structure that, for example, has been identified as promising for a particular application by molecular simulation.
We are developing tools that will allow to predict the formation of a MOF structure taking into account the synthesis conditions.
MOFs are synthesised in a self-assembly process from different building blocks: metal corners and organic linkers are combined
in a kind of molecular Lego set. This modular synthesis leads to a near infinite number of possible structures. Depending on
the synthesis conditions different structures can be synthesised even from the same building blocks making it difficult to target
a specific structure that, for example, has been identified as promising for a particular application by molecular simulation.
We are developing tools that will allow to predict the formation of a MOF structure taking into account the synthesis conditions.
Periodic mesoporous silicas
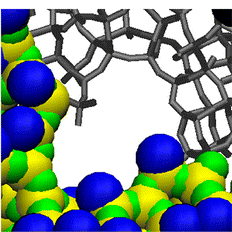 We are also developing models for periodic mesoporous silicas such as MCM-41 or SBA-2. These materials show long-range order
but are amorphous on the short range so that the detailed atomistic structure cannot be determined from experimental methods.
By mimicking their synthesis procedure, we have created realistic models that can not only be used to correctly predict
adsorption e.g. in carbon capture applications but also provide insight into the experimental solid. For SBA-2, for example,
we could show that their large spherical cavities are connected by windows rather than by channels as previously thought and
explain their origin.
We are also developing models for periodic mesoporous silicas such as MCM-41 or SBA-2. These materials show long-range order
but are amorphous on the short range so that the detailed atomistic structure cannot be determined from experimental methods.
By mimicking their synthesis procedure, we have created realistic models that can not only be used to correctly predict
adsorption e.g. in carbon capture applications but also provide insight into the experimental solid. For SBA-2, for example,
we could show that their large spherical cavities are connected by windows rather than by channels as previously thought and
explain their origin.
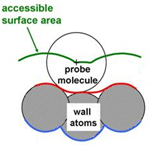 We use a variety of different geometric tools to characterise the porous materials we are working with computationally calculating
for example surface areas and pore size distributions. For crystalline materials where the positions of the atoms are exactly
known from experiments, this means that materials can be compared in a consistent way which is not possible to the same
extent with experimental methods based on e.g. adsorption isotherms where underlying assumptions might influence the result.
We have shown for example that the accessible surface area obtained by rolling a probe molecule along the surface provides
a good indication of the quality of a synthesised MOF sample when compared to the experimental BET area. The tool is freely
available under the creative commons licence and can be accessed
here.
We use a variety of different geometric tools to characterise the porous materials we are working with computationally calculating
for example surface areas and pore size distributions. For crystalline materials where the positions of the atoms are exactly
known from experiments, this means that materials can be compared in a consistent way which is not possible to the same
extent with experimental methods based on e.g. adsorption isotherms where underlying assumptions might influence the result.
We have shown for example that the accessible surface area obtained by rolling a probe molecule along the surface provides
a good indication of the quality of a synthesised MOF sample when compared to the experimental BET area. The tool is freely
available under the creative commons licence and can be accessed
here.