Workshop 3
Your assignment
A
healthworker has been accused of deliberately spreading multi-drug
resistant Staphylococcus
aureus through a ward in a general hospital. Three
patients
who were infected with MRSA are suing the Healthcare Trust for
substantial damages and they have turned to you for help.
Using
the skills you have learnt in the earlier workshops, decide whether you
think
the strain of MRSA found on the healthworker is the same as that
isolated from the patients. The marker protein we have used
is Clumping factor A (or ClfA) which shows significant variability
between strains. The DNA and protein sequences of ClfA from the three
cases are given below:
Having constructed your tree, submit it as part of a 1 page report (the Healthcare Trust are too busy to read anything longer), roughly one third of the page should be your introduction&methods, one third the results (the tree and associated legend), and the final third the discussion. As with the previous workshops, its the activity of producing your own tree that is the important part, so the report should not be long.
Hints:
- Do not use Internet Explorer or Edge.
- Find 12 to 16 more S. aureus
clumping factor protein and DNA sequences - this is instead of sampling the community for local S. aureus strains.
- Search
the databases by protein sequence as you want to find
sequences similar to those of the healthworker and the patients, so use
BLAST .
- You will need to do 4 searches, one for each of the healthworker and the 3 patients, using the ClfA protein sequences above.
- You will need to choose 3 to 4 sequences from each search but do
not
choose
just the top 4 for each search,
but a selection of those with the lowest E-value (this may be 0.0) and longest Query
coverage to 'sample the community'.
- Once you have chosen a protein sequence, click on the Accession code on the right-hand-side to take you to the Genpept page for that sequence:
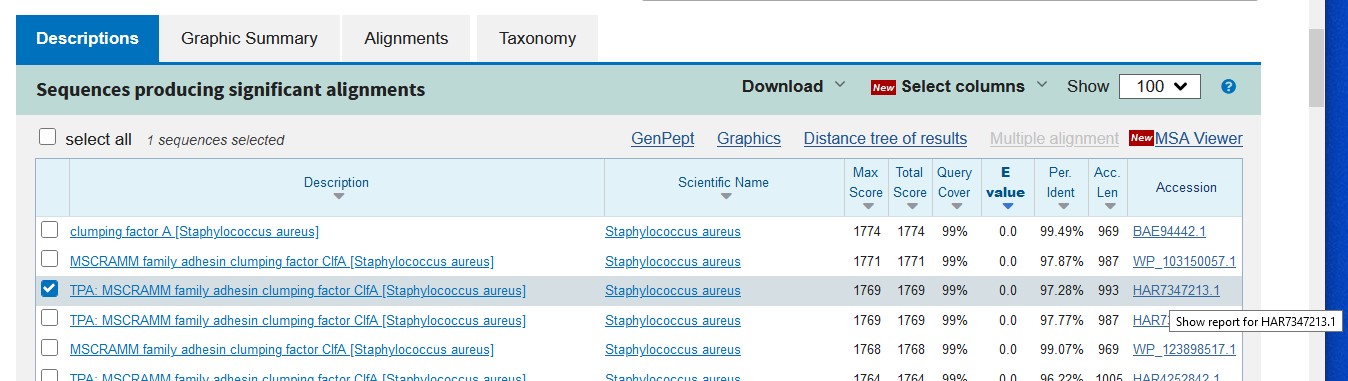
- change its format to FASTA via the link at the top of the page, and save it into a Notepad (or Wordpad or TextEdit)
plain text file.
- Find the corresponding DNA sequence. The simplest way is via a CDS link if you go back to the protein Genbank file (usually just above the protein sequence as shown here),
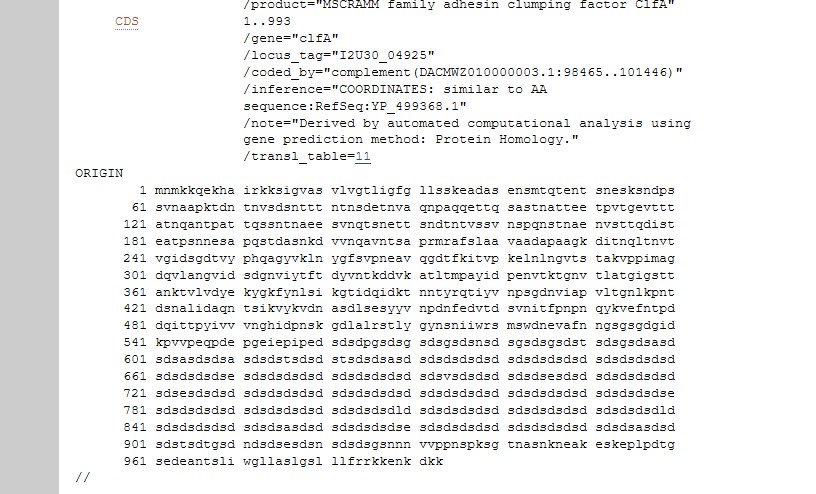 then by clicking on the FASTA link that appears at the bottom of the screen.
then by clicking on the FASTA link that appears at the bottom of the screen.
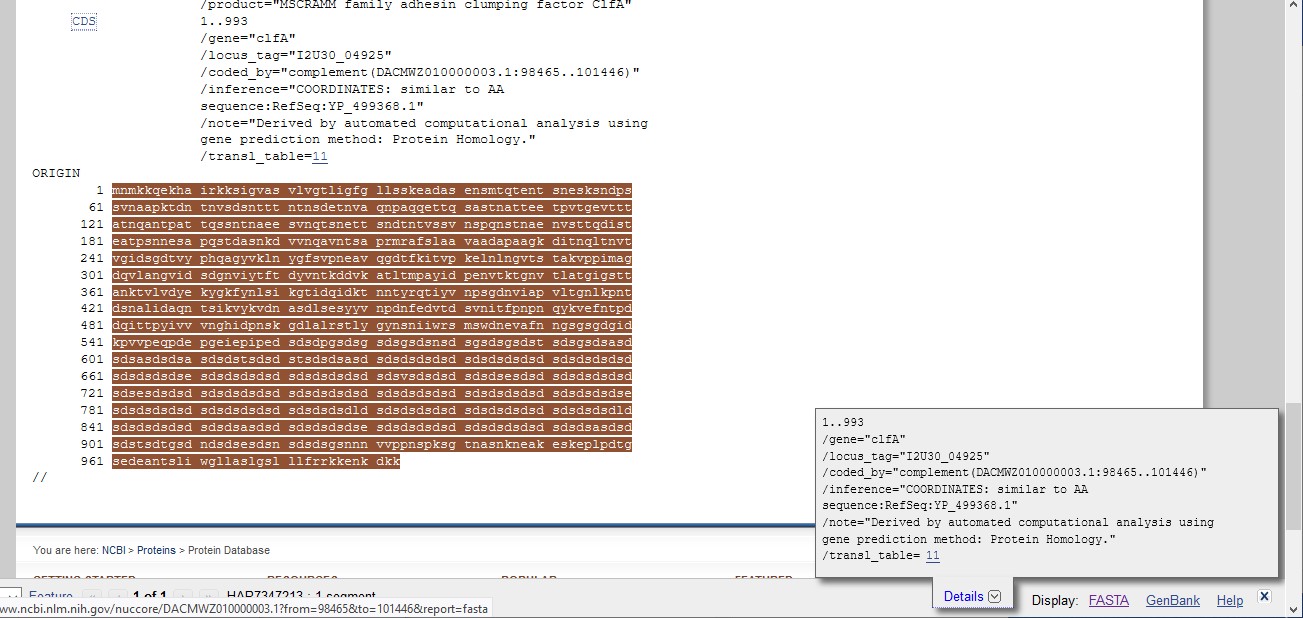
- If your chosen sequence doesn't have a CDS link, it should have a Nucleotide link on the righthand side of the page as shown below.
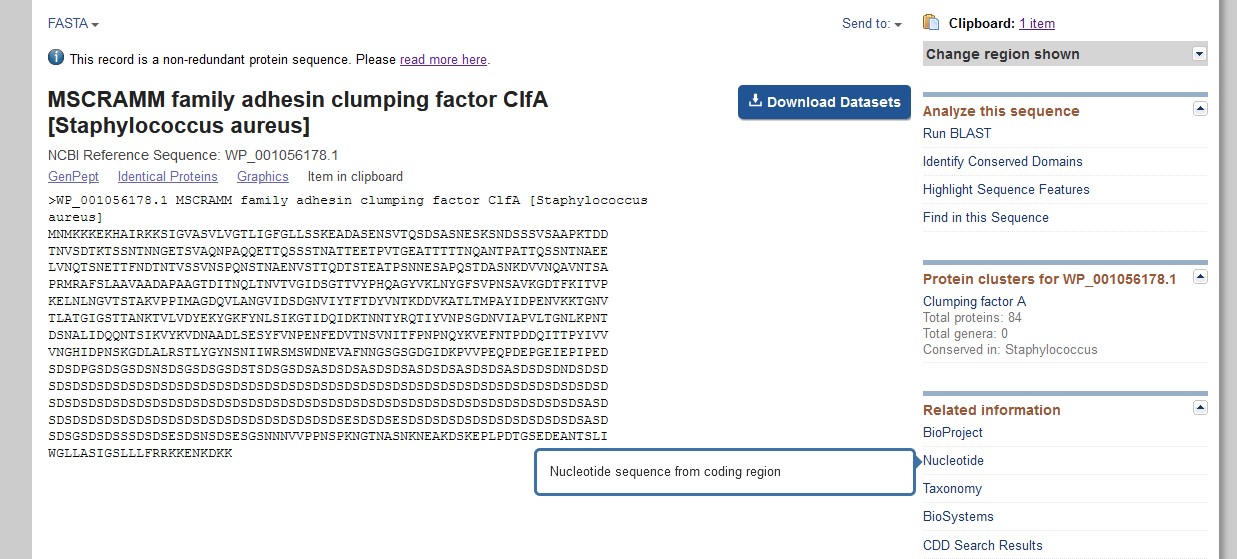
- The Nucleotide option may link to a number of sequences, if it does, choose the top one unless you have chosen it before.
- If your chosen link takes you to a complete genome sequence, but without any sequences visible, use the Customize view option on the right-hand side to Customize the view to show Gene, RNA and CDS features only as shown below.
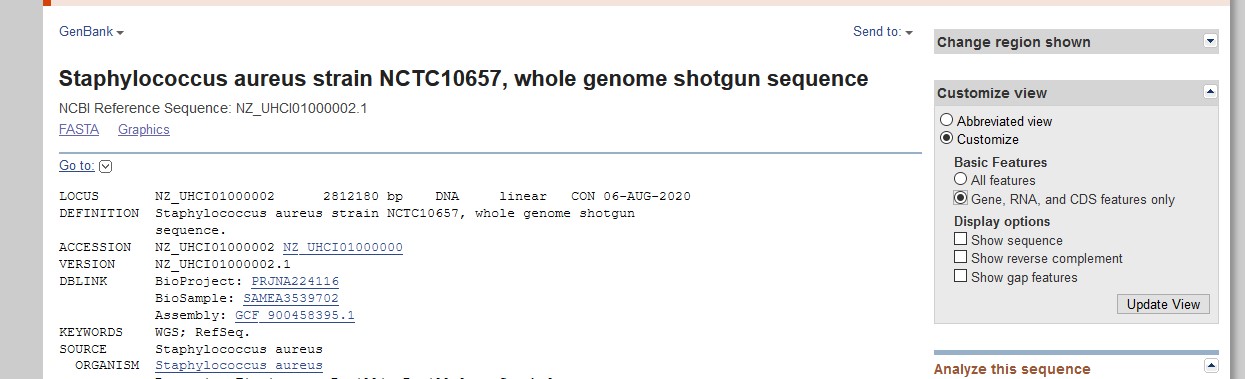
- Once you have some annotation to search, use your web
browser to find "clumping factor A" in the annotation and click the CDS
link on the left-hand-side (at the top of this image) to extract just the portion that corresponds to ClfA
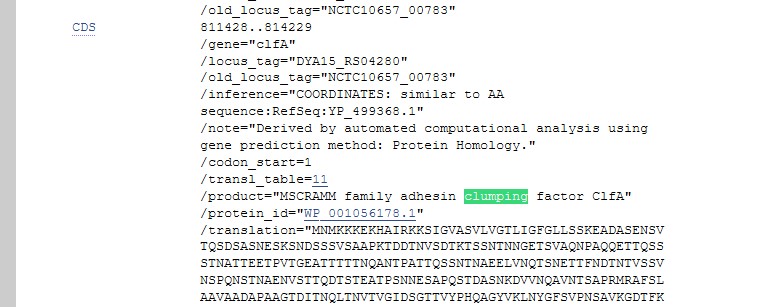
- Either you will be taken to the genbank entry for the sequence and may need to change this to FASTA to save it, or if a pop-up appears on the right-hand-side, (as shown below) click on FASTA just below it to get to the corresponding nucleotide sequence
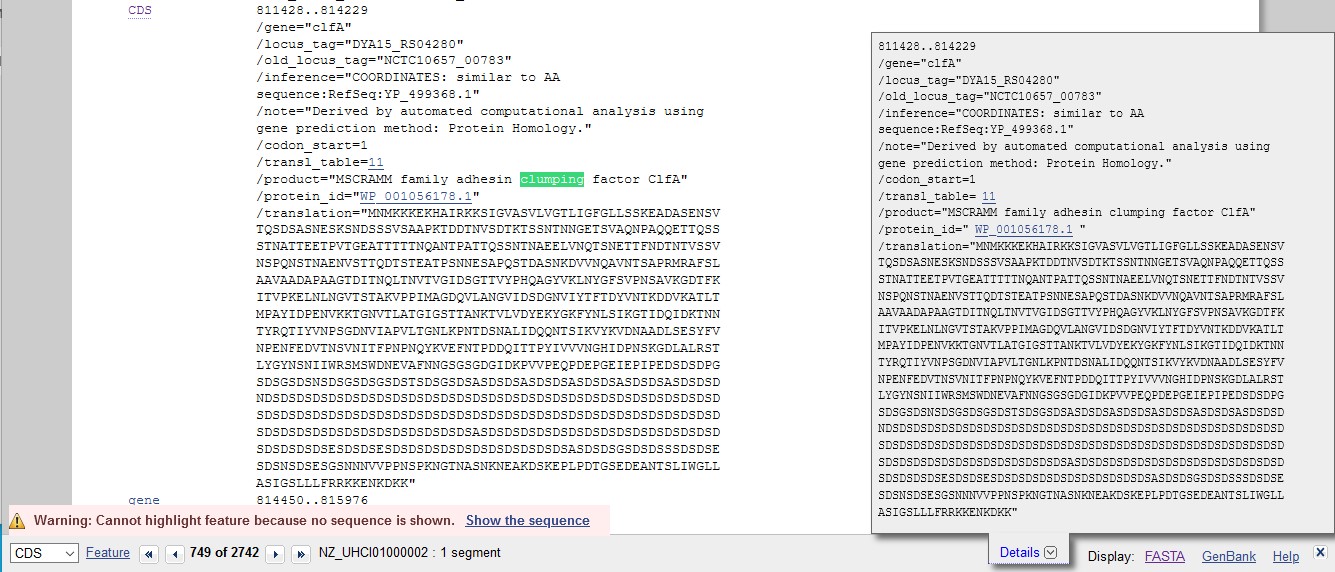
- However you arrive at the nucleotide sequence, save it in FASTA format (with its header line) to a file. Do not worry if the title still says whole genome, you can see by the length of the FASTA DNA that it is only a small part of the genome, (the portion between the bases in the box on the top right), the section that corresponds to Clumping Factor A.
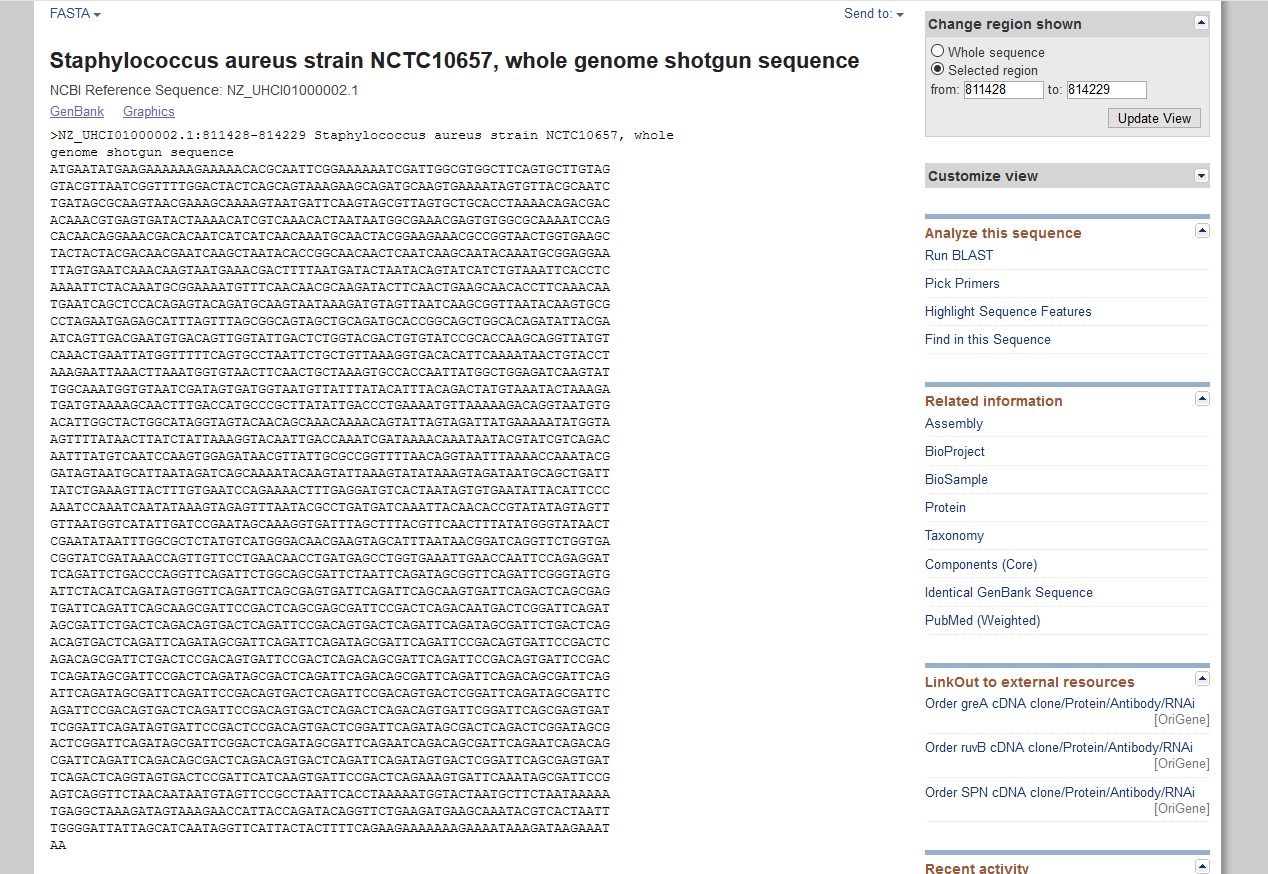
- Some sequences may be reverse transcribed. If your protein sequence starts with M, check that your DNA
sequence starts with ATG, and if it doesn't, check whether it ends with CAT (like the one below).
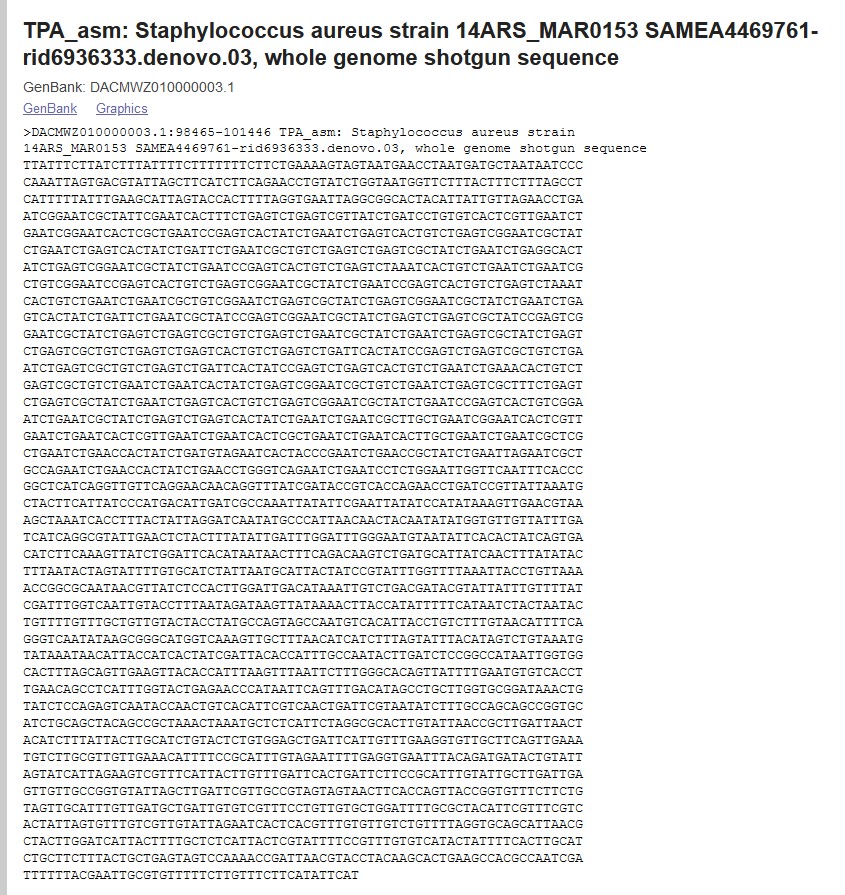 If so, you probably
need to obtain the reverse complement of your sequence using a tool
such as this. Put your sequence into the box and it will reverse complement it. Put this sequence into your growing nucleotide file rather than the original, although you should use the original header.
If so, you probably
need to obtain the reverse complement of your sequence using a tool
such as this. Put your sequence into the box and it will reverse complement it. Put this sequence into your growing nucleotide file rather than the original, although you should use the original header.
- Put
all the protein sequences (retaining their header lines too) in FASTA
format into one file, and all the DNA sequences (also in FASTA format, together with their header lines)
in another, making sure you keep the protein and DNA sequences in the
same order and that the files are in plain text
format.
- Once
you have your complete sequence selection, make sure you have no duplicates,
(this is easiest to do by looking for duplicates of the header lines). If you do, just delete
them (from both files), you only need 6 to 8 well varied ones, together with those of the Healthworker and 3 patients to draw the tree.
- Make a multiple
protein sequence alignment using T-coffee.
- When using Tcoffee, it
helps to keep track of the correspondence of protein to nucleotide sequences if you set the output
order to INPUT so that the aligned sequences remain in the same
order.
- Remember to include the
Healthworker and Patient protein sequences that are given at the top of this page, or they will not appear on the tree.
- If T-coffee claims there are 0 sequences to align, go back and
try to align only
2. If that works, add the others back one at a time until you find the
one that doesn't work. Remove that from both the protein and DNA files.
- Overlay the nucleotide sequences on the protein sequence alignment using
RevTrans as in workshop
2.
- no warnings should be printed to the screen,
any errors indicate that the
particular pair of protein and DNA sequences don't correspond to one
another.
- If you get an error message
about duplicates, check that you don't have any duplicate sequences. If
some of your sequences are short, it sometimes gets confused, but if
you move all the short ones to the end this often helps.
- Remember to output your aligned nucleotides in FASTA format.
- Make
a phylogenetic tree from the nucleotide sequences.
- You will have already downloaded SeaView in the previous workshop, so you don't need to do that again, just find the folder and run the executable there.
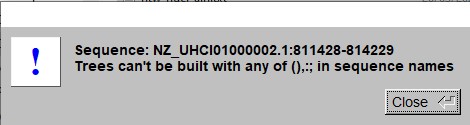
- If seaview complains about (),:; in the sequence names (as below), open the aligned nucleotide file and replace the : by a _ or similar. Then read the file in again.
- Only one tree is needed