In this exercise we'll start with the sequence of some DNA we have extracted from the malarial parasite, Plasmodium. We'll look for similar sequences in international databases to find out what the gene does. If it is essential for the parasite to live, it is a target for an antimalarial drug. Next we'll infer the molecular structure of the protein in order to begin the process of designing a drug to block it.
Steps involved in this workshop
Imagine you have obtained a DNA sequence such as the example sequence here. This file is in GCG format.
For the purposes of this workshop, we need to translate it because searching for similar sequences is more successful with proteins than with nucleotides. Why is this ?
We will start by using the software available at the European Bioinformatics Institute (EBI) so open another window at the www.ebi.ac.uk site.
All sorts of useful tools are available under the Services menu, click on this header at the top right of the screen, or Services > below the search box.
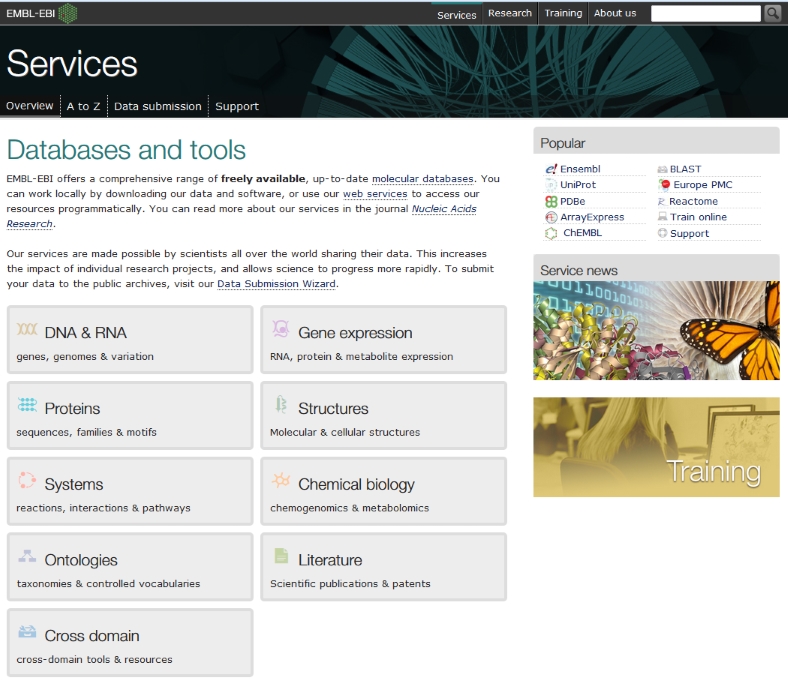
This brings up the main menu for the services at ebi, which are many. We have a DNA sequence, so click on DNA & RNA
Many tools are available, but the translation tool we need is found in the EMBOSS
tools. Scroll down and click on 
In order to translate this sequence we will use Transeq, one of the many
manipulation programs available.
Programs are available to translate nucleotide to protein, and to back-translate,
although this is less useful. Click on Launch Transeq .
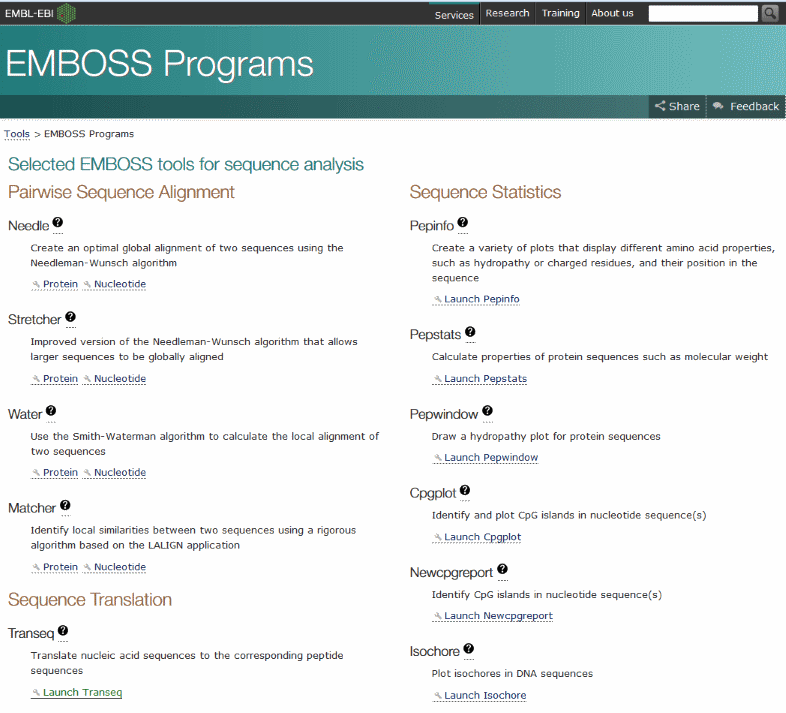
The result window shows a translated sequence containing many * characters, which mark untranslatable codons. This means that starting the translation at the first base doesn't use the correct reading frame to get a meaningful protein sequence from this DNA.
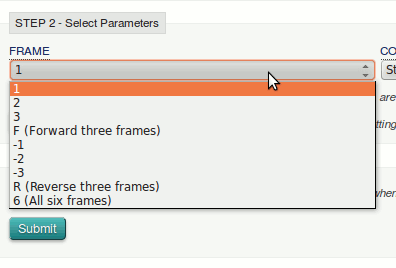
Here you can choose which protein translation frames to use, starting on the 1st, 2nd or 3rd base, all three forward directions (F), and the same in the reverse direction (-1, -2, -3 and R), or all 6 possible frames. We don’t know which is appropriate, so need to select the option 6.
Now click Submit
This time the output is a listing of 6 reading frames. Look through them for a long region without spurious stop codons (shown as *).
Both the 5'3'Frame 2 translation (unknown_2) and the 3'5' Frame 1 (unknown_5) fulfil this criterion, but the former (2) is most likely to contain the ORF, because 5 has a strange amino acid composition, very repetitive with amino acids often coming in pairs and even three V's in a row.
Having decided on the reading frame, go back to the Transeq window again, change the Frame to 2 and click Submit . Make sure the output looks reasonable (i.e. a long stretch of sequence without * or other abnormal amino acids).
Click on the Download tab above the sequence result. This opens a window containing just the sequence which can be saved to a file. Make sure when you do this that you save it as type Text File, or as All Files (i.e. not as an html file), and put it on your h-drive, (your University home drive), not this computer's desktop.
We need to edit this file so it contains just the open reading frame, i.e. from the first M following the first Stop codon, to the next stop codon.
Open the file in WordPad/Notepad. The sequence has been saved in FASTA format, this means that it starts with a header line beginning with a >, then has the sequence in single letter codes starting on the next line, listed without any spaces. On the sequence line, remove all the amino acids before the first M, and all those following the next stop codon * (including the stop codon).
Save this file with a name such as sequence.fasta, I've chosen this because it is a sequence file in "fasta" format, preferably with Type All Files.
Now we have our protein sequence we can use it to search for similar sequences. For this database searching we'll use the popular BLAST program. There are versions on the EBI website accessible from the Services page in the Popular section
but for variety we'll use the NCBI version which can be found at http://www.ncbi.nlm.nih.gov/BLAST. The BLAST program, searches the most up-to-date sequence database of using an array of computers in Washington DC.
The Web BLAST family of programs includes:
| Nucleotide BLAST | compares a nucleotide query sequence against a nucleotide sequence database |
| Protein BLAST | compares an amino acid query sequence against a protein sequence database |
| blastx | compares a nucleotide query sequence translated in all reading frames against a protein sequence database |
| tblastn | compares a protein query sequence against a nucleotide sequence database dynamically translated in all reading frames |
Below this there is the option to BLAST Genomes , that is to restrict your sequence search to a single species, which can be very useful if, for instance, you are working only on human genetics. However in this case we will leave this part alone and thus the whole database of all sequences will be searched with BLAST.
Since we have already determined the
correct translation of our nucleotide query sequence, choose
the  option.
option.
Click in the search box, below the Entry Query Sequence header and Paste your protein sequence into the box.
The box should now contain something like this:
>unknown_2
MPKAIVNVSGMHGGVMATLIVQKNTGDVVLFQIVKNMPHGKALDTSH
TNEMAYSNDKVSGSNWYDDAAGADVVIVTALFTKAKKSDKNWNRDGLLPLNTKSMIDIGP
HIKINCPLAFVIVVCNPDDVMVQLLHQHSGVPKNKIIGLGGVLDTDRLKYYISQKLNVCP
RDVNAHIVGAHGNKMVLLKRYITVGGIPLQEFINNKLISDAELEAIFDRTVNTAGEIVNL
HASPYVAPSAAIIEMAESYLKDLKKVLICSTLLEGQYDHSDIFGGTPVVLGANGVEQVIE
LQLASEFRKAFDEAIETKRMKALA
Leave the Database choice (under Choose Search Set) as Non-redundant protein sequences (nr) (from all databases).
If you were working on a particular species, you could limit the search to a particular organism in this section.
You can change any of the options or parameters for the search, such as the algorithm, the sensitivity of the search or appearance of the result, by clicking on the Algorithm parameters hyperlink at the bottom of the window. If you would like to understand all the parameters being used, clicking on the blue words on the BLAST page gives excellent help.
Click the  button to search the database for similar
sequences.
button to search the database for similar
sequences.
A new screen appears telling you your protein search has been queued, and the “request ID”. A little later it will also display any conserved domains identified through sequence similarity. If the system was slow you could save this and recover your results later.
This page is automatically updated until your results are ready...
When, at last, the results arrive, at the top of the page is the "conserved domains" chart originally shown on an intermediate page. Clicking on this chart gives more detailed information in a new window:
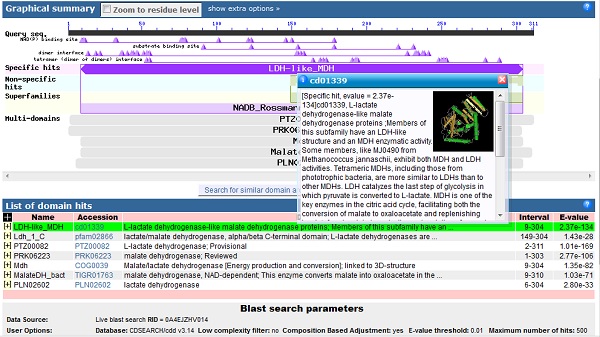
The uppermost black bar represents the sequence you submitted, underneath which the triangles represent important binding residues. Below these, the large purple bar shows that the sequence has a high level of similarity with the LDH-like MDH family, i.e. the L-Lactate dehydrogenase like malate dehydrogenase family. Hovering the mouse over any of the triangles or bars underneath gives more information. Clicking on the bars gives another screen with sequence alignments and trees of related sequences.
Having explored these, go Back to the main results page where
a graphical representation of the 100 most
similar sequences from the database are overlaid on our query
sequence. 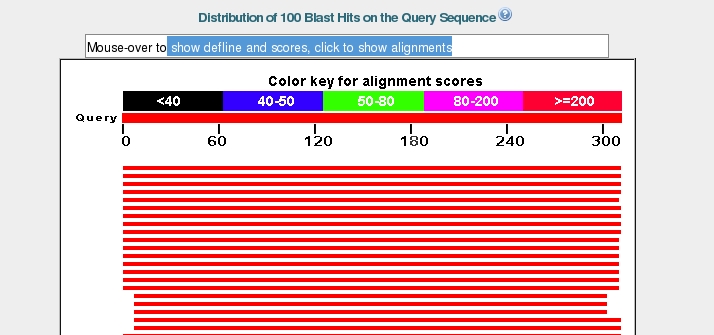
Again the top bar represents the sequence submitted, with the bars below representing matched sequences, coloured by the alignment score, with red being the highest. The length of the bar indicates the region over which there is similarity with the sequence you submitted. Hovering over a bar will bring its description up in the box at the top.
Below this chart come the names of the 100 similar database sequences (called "hits"), listed in descending scores of similarity.
The most important score to look at for similarity is the E-value, which describes the goodness-of-fit of a random sequence, relative to the hit. i.e. the lower the E-value, the better the fit of the hit to your sequence. A more detailed explanation is given here
Further down the results page are given the alignments of the database hits against the query sequence. It appears that our sequence is L-lactate dehydrogenase, which is essential for this anaerobic parasite to live, and therefore a drug target.
Going back up
to the Descriptions part of the page, choose
one of the proteins from the list. Clicking its
Accession hyperlink on the right takes you to the sequence file in
the database. If you wanted to save the sequence, a more useful format
is FASTA. At the top of the page for the sequence you have chosen, 
click on the Send to: link, then Choose Destination File, and finally choose FASTA in the pull-down menu. Save it in your area on the H: drive and change its filename to something more memorable.
Instead of BLAST we could have used the FASTA program to carry out the search. It uses an entirely different algorithm, so it's worth trying if BLAST doesn't give sensible results. If you want to compare methods later, FASTA is found on the EBI Services page http://www.ebi.ac.uk/services under Proteins.
To do this we want to limit our search to those sequences which have entries in the Protein (structure) Data Bank. This can be done using BLAST by changing the entry in the Database box so that the Protein Data Bank proteins (pdb) is searched. However it is more straight-forward to search the PDB directly. Click on http://www.rcsb.org/pdb. (This works better in a non-IE browser)
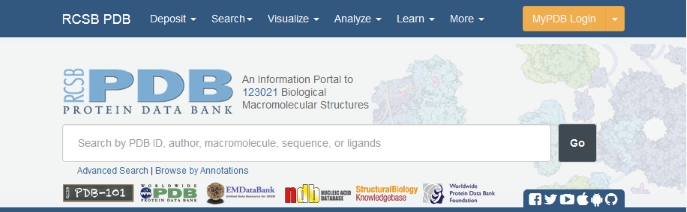
On the PDB site simple searches are carried out using the box at the top of the page. To do a search for structures similar to our sequence, copy the sequence into the search box, but this time without the header line.
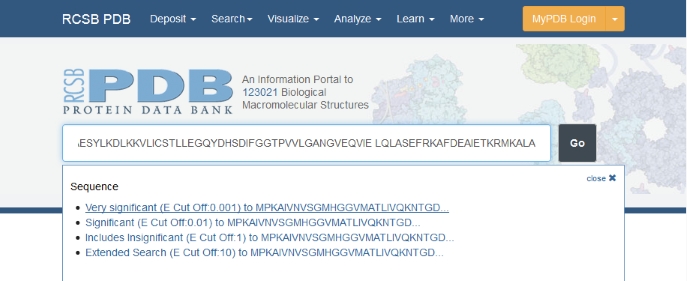
Another pulldown menu appears, asking which significance level you would like to use for the search. Choose the most stringent, you can always lower it if you get too few results. Then click the Go button.
The search results in a list of proteins of decreasing sequence similarity, (E-value), together with alignments between the query and database sequence, but this time the structure of each of the proteins is known. Choose one, e.g. 1T24, and click on it to bring up the entry in the PDB.
This page gives details of the structure, ligands bound, links to other databases, etc. However we want to view the structure. The RCSB PDB site has a number of programs available to view structures, hyperlinked from the Biological Assembly box on the left hand side. Click on NGL
Initially NGL displays the molecule as a cartoon showing the alpha helices and beta strands coloured from blue at the N-terminus through a rainbow to red at the C-terminus. Ligands are shown in space-filling representation. You can rotate the molecule by dragging the mouse with the left button pressed.
The appearance of the molecule can be changed using the right-hand pull-down menus, for instance you can emphasise the secondary structure by changing the Color scheme to By Secondary Structure.
3D structures of proteins are useful to design drugs to bind tightly to them, usually to inactivate them. There are more sophisticated programs than NGL, which allow you to build small molecules, which fit snugly into the active area of the protein. The protein may be a receptor in the human body that is over-responding to a hormone, or an activator necessary for tumour growth, or an enzyme essential for a virus to replicate. In each of these cases deactivation of the protein would help cure the disease.
The working sequence above matched sequences in the database very well, and it was pretty obvious what the protein was. Next, we'll do the same for a protein for which it is difficult to find any matching sequences, the sequence of which is here.
Go back to the BLAST (http://www.ncbi.nlm.nih.gov/BLAST) site and click on protein blast again.
Click in the sequence box, then Paste in the new sequence. If necessary, change the Database box back to Non-redundant protein sequences.
Then in the third,
Program Selection section, choose the PSI-Blast
algorithm then press .
The result pages are similar to those given by the ordinary BLAST. The first one shows that similarity has only been found to about the first 75 amino acids in the sequence. In the main results window, from the graphical summary it is clear that this level of similarity is much lower than that obtained with the LDH sequence above, the bars are coloured blue or black rather than red. From the scores and alignments, it is clear that BLAST couldn't find any homologous sequences in the database.
PSI-BLAST is just like BLAST, but is useful in finding homologous sequences with low similarity. If BLAST has failed to find a family of homologous sequences, try PSI-BLAST.
The way it works is it takes the sequence "hits" found in the first round of searching and overlays them to build up a matrix, containing account of what types of amino acids occur at each position. This is then used as the query sequence for the next round of searching, or "iteration".
Results of the first iteration are fed into the second, and those of the second are fed into the third, and so on, until convergence (the point at which no new sequences are found) is reached.
Thus it works on the principle that if sequence A is genuinely homologous to sequence B, but their similarities are not good enough to be spotted by BLAST, then if sequence C was found to be similar to A, then C might be successfully used to search for homologue B.
Choose some of the insignificant hits (select the boxes corresponding to your chosen sequences in the Select for PSI blast column on the right hand side) and run a second iteration using these by clicking the Go button below the sequences. The results now are more similar in sequence (the bars are pink or green). Continue running some of the iterations until convergence is reached.
It would improve your skill if you investigated a sequence by yourself without step-by-step instructions. You could pick up a sequence from the General Query (GQ) Sequence Search site (http://www.ncbi.nlm.nih.gov/gquery/). Type in the name of your favourite molecule and take a nucleotide sequence or go to Nucleotide and enter X16610 for a cytochrome P450 example.