
The structure and stability of solid surfaces are key factors that control a wide range of technologically important processes, including sintering, catalysis and corrosion.
 Modelling Real Surface Structures
Modelling Real Surface StructuresEnergy Screening - Hematite
In nature surfaces are not totally flat and defect free.
Therefore we must sample all possible configurations. In this example we
consider the Hematite (01.1) surface and decribe a process called energy
screening.
< align=left>The hematite (01.1) unit cell consists of 20 ions. There are 2
20 possible configurations. By considering non dipolar surfaces and
symmetry this is reduced to give 1277 reasonable configurations.>
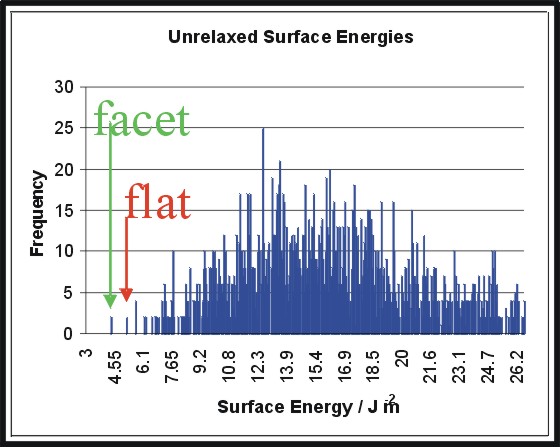
Each surface configuration is relaxed. We find that many of
the most stable surfaces are facetted, for different materials.
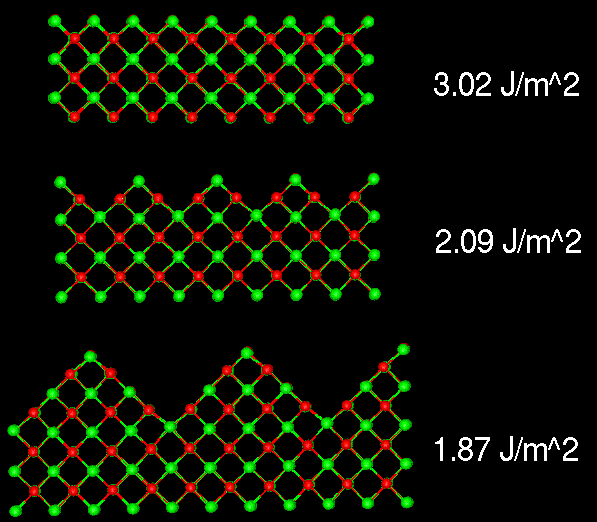
Flat, 3.0 J/m2
Facet, 2.6 J/m2
Microfaceting on the {110} surfaces of MgO.
(25K) |
related references:
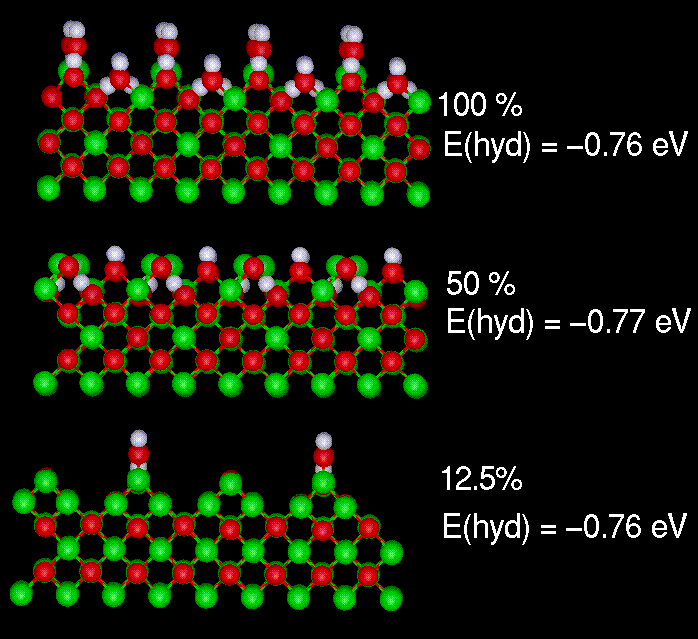
The hydration energy for the faceted {110} surface of MgO was calculated
as a function of coverage. The energy is essentially constant showing classic
Langmuir behaviour.
Hydration energies for the {110} surface of MgO.
 (46K) |
related references:
The average co-ordinates for the 20ps data collecting run for {110}
and Ni {111} slabs indicate that there is considerable disorder at
the surface. This is especially evident for the Ni {111} surface
where the diffusion constant for the ions in the top most layers
approaches that of a liquid at this temperature.
An interesting result from the {110} simulation is that even when
the flat surface is modelled it rearranges to form a facet structure
similar to that described above for MgO.
Molecular dynamics simulation of the {100}, {110} and Ni {111}
surface of NiO at 2000K.
 (31K) |
related references:
Computational solid state chemistry group home page
![]()
page design by Graeme Watson and Pete Oliver.
Send your comments and questions to
s.c.parker@bath.ac.uk.