
NiO is isostructural with MgO and whilst the crystal morphology of MgO is
cubic NiO is not. The surface energies for the {100}, {110} and {111} surfaces
of NiO were calculated but the ulting equilibrium orphology
(using a Wulff construction) was dominated by the {100} ie cubic. However,
the discrepancy can be explained by appreciating that NiO is easily oxidised
and hence the surface energy as a function of surface oxidation was calculated
for all three surfaces. It was found that at 75% oxidation of the surface the
calculated morphology agreed well with the experimentally observed
morphologies. Thus we have shown that oxidation can stabilise an otherwise
unstable surface ie the {111} surface.
 Computer Simulation of the crystal morphology of NiO
Computer Simulation of the crystal morphology of NiO
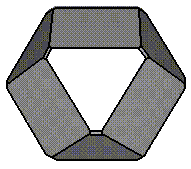
Experimental Morphology |

Unoxidised |

75 % Oxidised |
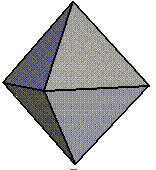
100% Oxidised |
related references:
Computational solid state chemistry group home page
![]()
page design by Graeme Watson and Pete Oliver.
Send your comments and questions to
s.c.parker@bath.ac.uk.