In this exercise, we'll start with a set of gene sequences coding for the same protein in different organisms, and align them. Two methods of presentation and highlighting significantly homologous areas will be evaluated. Then we'll use the alignment to calculate trees using various methods, which may throw light on the evolutionary relationship between the organisms to which these genes belong.
4. Nucleotide sequence alignment
First we need to find some sequences. In the last workshop we searched for sequences by similarity with "our" sequence, using BLAST. However each sequence in the databases will also have been "annotated" with anything that is known, or can be inferred, about its function. This time we'll search the annotation for proteins with a desired function. This method of searching uses a different www interface to the databases, the Global Cross-database NCBI Search or GQuery (formerly Entrez).
When you have arrived, enter
in the box and press the Search button. When you do this for real you'll put the name of your favourite protein here.
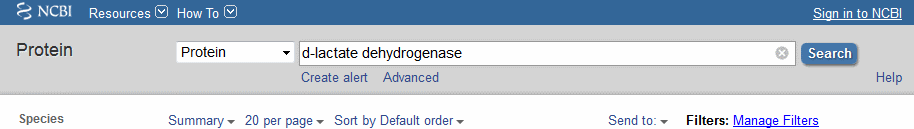
The results page shows hits in almost all the databases, including, further down the page, more than 65000 protein hits. Click on this row to bring up a Protein Results page (like that below).
Select, using the square buttons on the left, five or six sequences (avoid sequences that appear to be from the same species and those only “dehydrogenase-like” or hypothetical). At the top of the page change the Summary to FASTA (Text). The view will change to show only the chosen sequences in the FASTA format.
You may also wish to have a sequence for which the structure is known. These can also be extracted via GQuery, but we'll search the Protein Data Bank or PDB directly by opening http://www.rcsb.org/pdb. Here type
d-lactate dehydrogenase
into the Search box, and click on Go.
There are far fewer results than obtained from the protein sequences, only about 185, and even this small number includes all entries mentioning the words D-lactate dehydrogenase rather than all 185 being D-lactate dehydrogenase structures.
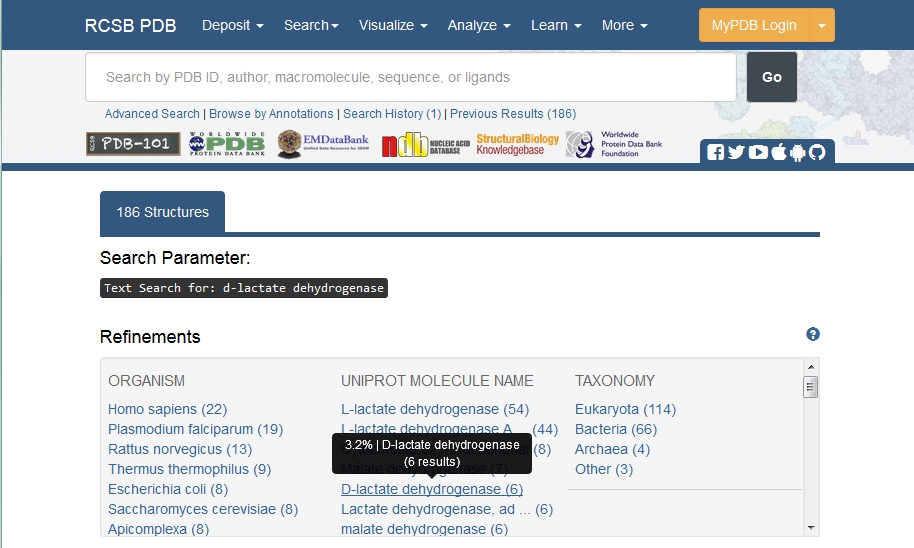
To retrieve only D-lactate dehydrogenase structures, click on the link under the UNIPROT MOLECULE NAME header.
Now there are only 6 structures, clearly illustrating the relative difficulty of obtaining structural information.
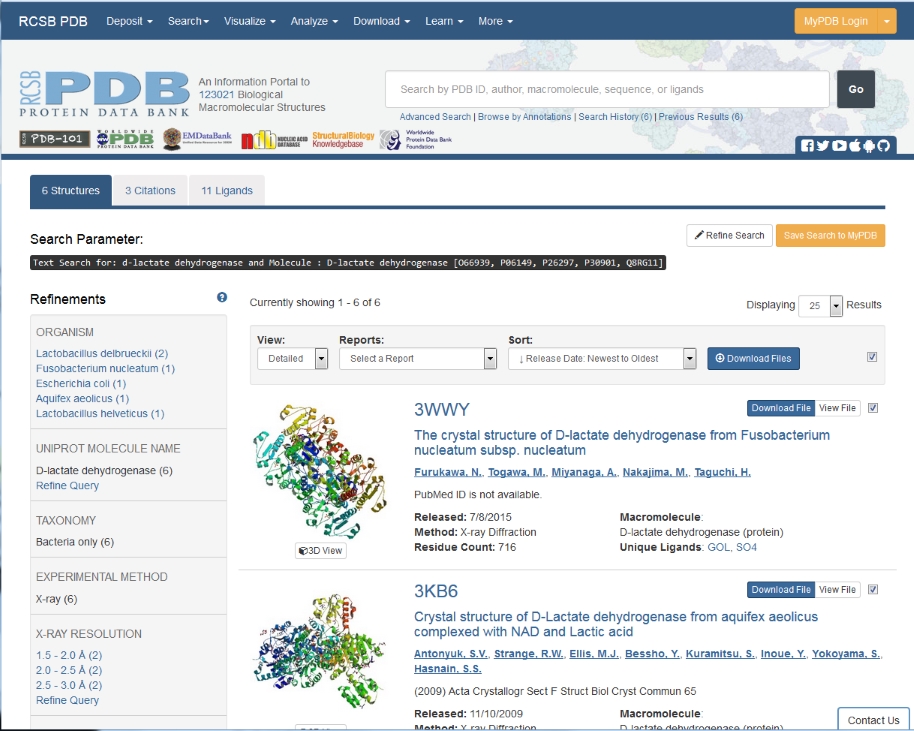
In the next window there is more information about the structure, as we saw in workshop 1, but for this exercise all we need is the sequence, so click on
 Download Files in the right-hand
toolbar, then on FASTA sequence.
Download Files in the right-hand
toolbar, then on FASTA sequence.
If you are asked whether you want to Open or Save the file, choose Open. In this case a Notepad window will open containing the sequence. Using other browsers you may need to click on the downloaded file. The sequence may appear to be all on one line, in which case you will need to edit it to put the header on a separate line by putting a line break (pressing Return) after the part that begins:
>1F0X:B|PDBID|CHAIN|SEQUENCEDepending on which PDB file you have chosen, there may also be more than one copy of the sequence, as there are more than 1 copy of the molecule in the crystal structure. 1F0X for instance has two, so there are 2 identical copies of the sequence in the file. You'll only need one of them and should find the second copy (also starting >1F0X) which will be somehere in the sequence text and delete from that >1F0X to the end of the file
Now you should have two windows, one Browser window with 5 to 6 sequences from your Entrez search and the Notepad window with a PDB sequence.
Many programs have been devised that attempt to produce an optimal multiple sequence alignment. Different programs are optimised for different sizes of alignment, and many are freely available on the web. To align the sequences we’ve collected, look at the options available from the European Bioinformatics Institute www.ebi.ac.uk.
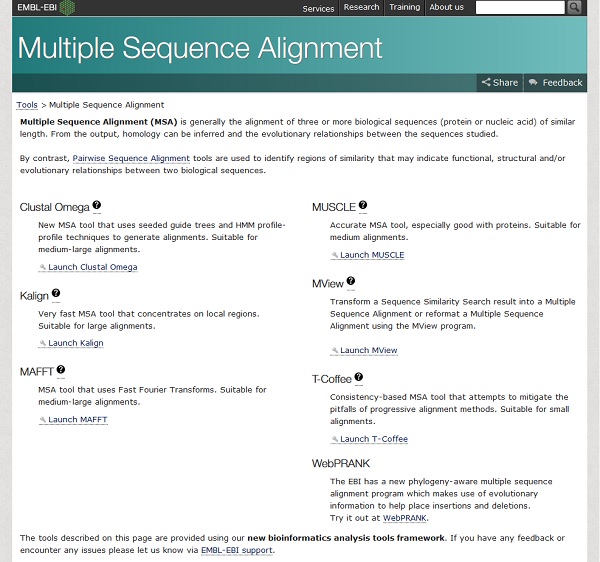
As we have only five or six sequences, this classes as a small alignment, so either MUSCLE or T-Coffee would be suitable, we will use T-Coffee.
Clicking on the Launch T-Coffee link takes us to a submission form.
Under STEP 1 - Enter your input sequences, copy each sequence into the sequence box. First the PDB sequence from the Notepad window, beginning something like
>1F0X:A D-LACTATE DEHYDROGENASE
Then copy the NCBI - Protein sequences, highlighting each sequence in turn, on the window including the header line beginning
>gi|
and use Edit, Copy, then Edit, Paste into the T-Coffee window.
(Hint: it is easiest to do this from a plain text file. At the top of the window change the Send to box to File and open the subsequent file to copy the sequences all at once.)
Scroll back up the box to check that the title line of each sequence is separated from the previous sequence, but that there isn't a blank line between the sequence and its header.
The sequence box now looks something like this
>1F0X:A D-LACTATE DEHYDROGENASE
MSSMTTTDNKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADK
IILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHS
VIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDR
IKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFY
IGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAEKYGKDTFLMIDKLGTDKMPFFFNLKGRTDAM
LEKVKFFRPHFTDRAMQKFGHLFPSHLPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFF
VCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHF
MCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGK
TSKRKNWQEVE
>gi|5123869
MTFSSTSSAPPPSPLLPATRITVYGCGRDEAALFRRTAPRFGVEATLTEAAVSEENAEMAAGNQCISIDH
KTPVTPATLRALHRAGVTYISTRSIGYNHIDVTYAAGVGISVENVTYSPAGVADYTLMLMLMAVRNAKST
VRRAELHDYRLNEIRGKELRDLTVGVIGTGRIGAAVVDRLRGFGSRVLAYGKRPTIAADYVSLDELLRSS
DIVSLHVPLTPDTHHLLDQSRIRRMKSGAFVINTGRGPLIDTEALVPALESGRLSGAALDVIEGEEGIFY
ADCRNRTIESTWLPRLQKMPNVLISPHTAYYTDHALMDTVENSIINCLNFGSRKQHG
>gi|5104136
MARIAEELEKIFGPEKVVSDPHIVRLYSREPSGLEGRAEAVVFPESAQDVSRLVRYAYSREVYIYPQGSS
TDLAGGAFPERPGVVVSMERMRRVREVSVLDSVAVVEPGVRLWDLNVELSKYRYMFPIDPGSVKVATVGG
AINTGAGGMRGARYGTMRDWVLGLEIVLPDEEGTILRVGCRTLKCRQGYDLARLIVGSEGTLAIVTEAIL
KITPMPENVVVVLAFFPTLRQLVDAVIE
>gi|624075
MVDSIISYATYGKMFSRTSPKPVMPKNLKPQVAIFSAGNYVKDFIKPIESICTPVYIESSLNETTAALAN
KCDAINAFVNDDLSAPVLDILKNCGVSSITLRCAGFDRLDIEYAKELGFNVYRVPAYSPRSVAELALTHM
etc.....
At the bottom, click Submit
Eventually a T-Coffee Results window will appear. (If no results appear and the subsequent window claims that 0 sequences were submitted, then return to the submission page, and press return at the end of each of the header lines beginning
>gi|
then resubmit.)
On the Results window, click on the Download Alignment File button. On the next pop-up window Save the file, changing the name to e.g. dldh.aln, the file type to All files,(if necessary) and save it to your H: drive.
We’ll look at various methods for presenting alignments that emphasise homologous sequence positions according to different criteria.
An applet version of JalView is available on the T-Coffee Results Summary page.
However as of 1st September 2015 the BUCS browers don't support Java, so we'll have to move on to the next section. I've left the information on how it works in case you can access Java on your own computer.
Click on the Start JalView box.Using this applet you may
Practise doing some of these things. A more powerful editor, which has to be downloaded to your lab pcs (BUCS don't have it) is BioEdit. If you need greater functionality in the future, then you can download it from http://www.mbio.ncsu.edu/BioEdit/bioedit.html . The edited file can be saved in a number of different formats (although you don't need to now).
Boxshade is a simple web-based program that presents a sequence alignment shaded by residue type. Click on http://www.ch.embnet.org/software/BOX_form.html.
At the top of the Boxshade window change the output format to RTF_new. In the bottom portion, chose your input sequence format, ALN.
Open the dldh.aln file in Wordpad (All programs > Accessories > Wordpad). Click Select all under Editing in the top menu, then Copy. Then paste into browser window.
Press Run BOXSHADE
This should produce a results window in which you need to click on
Output number 1
to obtain a RTF file of your alignment. The output may be displayed automatically, otherwise you can open it in Word after saving it to e.g. dldh.rtf. It looks better if you change the Page Layout to Landscape (Page Layout, then Orientation).
The www site for this program is http://espript.ibcp.fr/ESPript/ESPript/.
Here click on 
At
the top of the site it warns you to allow it to open
pop-ups on your screen. To do this, (in Internet Explorer) select Tools, then
Internet Options, then
First scroll down to the Secondary structure box,
and click on the ![]() symbol. In the new window, type the PDB ID for the sequence you
selected in part 1, so if you chose 1F0X as in the example, then type 1F0X
here and press Retrieve data
.
symbol. In the new window, type the PDB ID for the sequence you
selected in part 1, so if you chose 1F0X as in the example, then type 1F0X
here and press Retrieve data
.
If, as in the case of 1F0X, there are more than one protein chain in the structure, put the identifier for the sequence you chose, so it might be A, in the Chain ID box. Knowing the structure will result in a plot which annotates the sequence alignment with the secondary structure.
Now move back up to the Aligned sequences box and click the Browse button to search for the dldh.aln file.
If you are familiar with similarity matrices you can change them in the Similiarity calculations box, otherwise use the defaults. Change the output layout to Landscape with 110 columns (Col). By default the output will be as a postscript or pdf file, but you can specify other formats which are more readily inserted into Word documents.
Click on  on the top toolbar. A link will appear in a new RESULTS
window, clicking on the PDF link in which
should show you your output, the sequence alignment with the 1F0X
secondary structure represented diagrammatically at the top and
the alignment coloured by conservation. If your browser is set not to
allow
pop-ups, change your preferences for this session and re-submit your
data.
on the top toolbar. A link will appear in a new RESULTS
window, clicking on the PDF link in which
should show you your output, the sequence alignment with the 1F0X
secondary structure represented diagrammatically at the top and
the alignment coloured by conservation. If your browser is set not to
allow
pop-ups, change your preferences for this session and re-submit your
data.
Rather than protein sequences, it is better to use DNA sequences for phylogenetics because protein sequences have no information on silent nucleotide mutations. For successful molecular phylogenetic analysis, the sequences must be well aligned. However in general, it is better to use protein sequences for multiple alignments, because nucleotide sequences have only four bases, making many alignment possibilities and confusing the alignment program. So the strategy for making aligned nucleotide sequences is:
(i) obtain the nucleotide and corresponding protein sequences.
(ii) align the protein sequences
(iii) take the original nucleotide sequences and overlay them onto the protein sequence alignment
Obtaining all these sequences is too time-consuming for this workshop, so two sample files (unaligned nucleotide, and aligned protein) have been prepared for you.
(i)The protein sequences were downloaded into one file, in FASTA format, using GQuery as we did in step 1, then aligned and saved to myprotaln.aln.
(ii) The nucleotide sequences corresponding to each of the protein sequences were downloaded by following the links from the protein pages, and saved into a file mynuc.fasta,
We will use RevTrans to do the DNA alignment at this website http://www.cbs.dtu.dk/services/RevTrans/ .
Copy the nucleotide and aligned protein sequence from the links above into the appropriate boxes on the RevTrans website.
Further down the page, under the Advanced options header, change the Output format to FASTA for compatibility with the phylogenetics program. Then press Submit query
When the results page appears, click on Download result
 and save the subsequent file
to your h: drive as e.g. mynucalign.fasta. As this isn't always reliably saved as
plain text, it is a good idea to copy the text and paste it into a new Notepad file aswell,
saving it as mynucaln2.fasta.
and save the subsequent file
to your h: drive as e.g. mynucalign.fasta. As this isn't always reliably saved as
plain text, it is a good idea to copy the text and paste it into a new Notepad file aswell,
saving it as mynucaln2.fasta.
to make the phylogenetic trees we will use the molecular phylogeny program SeaView
This can be downloaded from http://doua.prabi.fr/software/seaview.
Click on the appropriate download for your computer, selecting Save when asked. Once downloaded, you are offered the chance to Run the programme, and extract it to a suitable location on your H: drive.
Run the program from that folder
Start the tree-building program, SeaView.
From the File menu, use Open FASTA to open the aligned nucleotide file you saved earlier.
There are three methods for Tree Building offered by SeaView.
First, Parsimony which uses PHYLIP to return the consensus of the most parsimonious tree found.
Second, Distance methods . Distance methods use attributes of the sequences to calculate the evolutionary distance between all pairs of sequences in the set. This relies on a definition of the mechanism of evolution i.e. a model for the mutation of nucleotides over time.
Third, the method of PhyML (Maximum Likelihood) is computer intensive, but uses the highly unbiased Bayesian statistics to get a more reliable result. This requires another download, so we won't be using it today
Other terms you will encounter include:
Bootstrapping which involves randomly resampling the data used to create the tree, in order to place confidence limits on the position of each subtree. 86% means that in 86 out of 100 resamples, the subtree fell in the same place.
Jumble is used in Maximum Parsimony and Maximum Likelihood, because these methods are dependent on the order that the sequences are listed. Jumble causes the calculation to be carried out several times in different orders, and chooses the best resulting tree.
In tree pictures, the distance between the branches mean nothing, it is the distance along the branches which describes the evolutionary distance between the sequences. The distance between any two sequences is the sum of the branch lengths connecting them, given in arbitrary units.
From the Trees menu, select Select Distance Methods . In the new menu select Bootstrap then click on Go.
In the new tree window, click on Br Lengths (Branch lengths) and Bootstrap. These give an indication of the distance between sequences, and the reliability of the branch points respectively. The result can be saved using the File menu to a PDF file, or using the (Print Screen key) and pasting the clipboard into a Word document.
Try the Parsimony method (under Trees) for calculating a new tree and compare results.